SMA
」 SMA 脊髓性肌肉萎縮症 健保 衛福部 健保署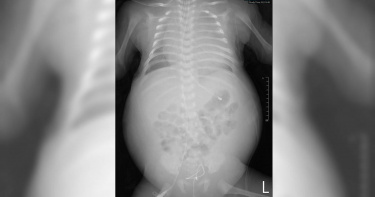
為保險延誤新生兒篩檢 27天大嬰兒險喪命
近一、二年來,約有2~3成新生兒父母,為了購買新生兒終生保險,在寶寶剛出生後只先接受法定的新生兒篩檢,約一至三個月左右核保通過後,再進行較全面的自費篩檢,這個觀念錯誤的情況,近幾年廣為流傳於家長社群中,但如無法即時篩檢出如龐貝氏症、脊髓性肌肉萎縮症(SMA)…等罕見疾病,可能錯失黃金治療時機,導致終生不可逆的傷害,甚至危及生命。台中一名出生27天的嬰兒王小弟,出現明顯的呼吸困難,緊急就醫後發現心臟異常肥大,肌張力低,肌肉酵素增高,高度懷疑為龐貝氏症,需插管治療。臺北榮總兒童醫學部楊佳鳳主任接獲通知後,立即派出醫療團隊,將病童接至臺北榮總,啟動緊急處置。雖到院當日立即完成診斷及給藥,但王小弟的病情仍然不樂觀。出現急性氣道壓迫,緊急進行「軟式支氣管鏡檢查」,立刻換置氣管內管,避免下端氣道完全壓迫無法進氣。在新生兒加護病房內,又頻繁出現心律不整,小小的胸腔被異常肥大的心臟塞滿,導致血液無法回流,合併全身水腫,心跳更降至每分鐘30下,情況十分危急,必須立即進行心臟按摩急救,新生兒葉克膜小組已在旁待命,經醫療團隊通力合作,成功挽救生命,住院治療40天,目前已康復出院多,未來僅需要每2周回來門診治療即可。龐貝氏症(Pompe disease)是一種罕見的遺傳性神經肌肉疾病,屬於體染色體隱性遺傳疾病。致病主要原因是溶小體一種酸性葡萄糖苷酵素(GAA)基因發生突變,導致GAA酵素無法分解肝醣,堆積的肝醣逐漸使肌肉受損,最後影響心臟和呼吸道肌肉。龐貝氏症疾病分成兩種類型,其中嬰兒型的患者若不及時治療,會在1歲前死於心臟衰竭或呼吸衰竭。在臺灣平均每2萬5千名新生兒就有1位是龐貝氏症患者。目前治療方式為每2周,施打病人體內所缺乏的葡萄糖苷酵素,越早治療預後越好。目前接受國內新生兒篩檢確診接受治療的約有100位病人。國民健康署補助21項新生兒篩檢,包括葡萄糖六磷酸鹽脫氫酶缺乏症G6PD(俗稱蠶豆症)…等等,詳如附表,惟不包含龐貝氏症、脊髓性肌肉萎縮症……等重大罕見疾病。王小弟的媽媽表示,「我們沒有做新生兒採檢,是因為剛出生的時候,我想要先為小孩規劃保險的部分,那等投保成功之後再去做自費篩檢,我沒想到會剛好,寶寶罹患龐貝氏症。」王媽媽表示,寶寶住院時甚至一度接到病危通知,幸好最後他挺過來了,真心呼籲大家「小朋友出生後盡早為小朋友做新生兒自費篩檢,這樣子小朋友跟爸爸媽媽,就比較不會需要承受驚慌和這麼煎熬的過程。」臺北榮總兒童醫學部牛道明主任呼籲每一位新生兒的家長,所有新生兒公費與自費篩檢項目,均經由專家學者反覆討論後制定,都是非常重要的疾病,尤其是龐貝氏症、脊髓性肌肉萎縮症(SMA)兩種罕見疾病,有著必須早期診斷、早期治療的急迫性。請各位家長務必依照正常程序篩檢,勿存僥倖心態,才不會延誤寶寶的治療,對長期預後造成重大影響。

罕病SMA藥物全給付 至今近200人受惠 病友迎重生奇蹟
衛福部健保署今年8月全給付「脊髓性肌肉萎縮症(SMA)」背針及口服藥物,不少病友終於等來重生奇蹟。根據健保署統計,截至今年到9月底,國內共有195名患者使用健保藥物,其中1人就是生命之窗慈善協會理事長李怡潔。接受了4次健保治療的她,運動功能已明顯進步,21日舉辦感謝宴,感謝健保署長石崇良給他們意想不到的支持,讓病友看到了不一樣的健保署。生命之窗慈善協會21日舉辦「重獲新生感謝宴」,李怡潔表示,自己體會過SMA無藥可醫的絕望、見證過治療奇蹟的發生,也終於讓幾乎所有SMA病友取得了救命藥。在倡議過程中,有部分病友因等不及而永遠地離開,令她深感心痛與遺憾。石崇良表示,多數的罕病病友無法選擇自己的人生,SMA的藥物提供了改變的機會,但藥物相當昂貴,一般家庭難以支付。健保署2020年開始支付SMA用藥,當時一年的藥費要500~600萬,只有有限的病友可以用。去年4月開始放寬,並在8月通過第一個基因治療的藥物,每針要價4900萬。「大家覺得該不該?我也在問自己這個問題」。石崇良反覆思考,4900萬的藥,健保付不付得起?但轉念一想,台灣有2300萬人,每人出2塊,不就可以買一支?這一支藥,可以改變一個人、一個家庭,更重要的是,讓大家對未來充滿希望。科學進步的結果,就是改變人生和命運。到了今年8月1日,SMA的藥物幾乎全給付,按照仿單,開放病友使用,他向全國的付費者表達感謝。對於今年8月全給付SMA背針及口服藥物,李怡潔感性地說,「謝謝署長給我們意想不到的支持,讓我們看到不一樣的健保署」。她姊姊也代她向大家深深鞠躬,期盼有更多SMA病友接受治療後,能改善病況,創造更多生命的奇蹟。

立委陳昭姿再爆衛福部「還有3司長」涉霸凌 臉書曝基層致謝:謝委員主持公道
近日發生的勞動部勞發署北分署公務員霸凌輕生案,成為引爆公部門基層人員反抗職場霸凌的火苗。繼海科館、疾管署、北市社會局陸續爆出職場霸凌事件後,有立委表示近日陸續接獲霸凌投訴,衛福部內恐怕還有「至少3名司長」涉及職場霸凌,點名衛福部長邱泰源稱,「不要袒護,要加油趕快查。」根據《ETtoday新聞雲》報導,民眾黨籍立委陳昭姿今(21)日上午在立法院衛環委員會質詢時,出示匿名粉專的文章,質疑衛福部主任秘書兼社保司司長劉玉娟有霸凌行為,劉玉娟立即否認回應「這不是我」,衛福部部長邱泰源也表示被指控者的背景與劉玉娟不符。事後,陳昭姿在SMA病友活動中受訪解釋,劉玉娟案件是民眾直接陳情,和粉專文章不是同一案件。陳昭姿宣稱,陳情人指劉玉娟是「長期霸凌案件」,在她到衛福部任職之前,還在健保署台北業務組時期就發生。陳昭姿抱怨邱泰源上午面對質詢,馬上用「工作認真」來回答,但「工作認真」和「有沒有霸凌」根本是兩件事,認為邱泰源不應該袒護。陳昭姿指出,就她目前掌握,衛福部至少有3名司長涉及霸凌,除了社保司的陳情,另有粉專貼文揭露的司長,其他委員辦公室透露另一名司長也涉及職場霸凌,還指出:「很遺憾,我目前看到都是女性。」陳昭姿今早揭露臉書粉專貼文發布於2023年6月,指稱該名司長在外標榜「年輕優秀」,實際上是霸凌下屬的無良長官,不分平日、假日,以口頭或Line交辦工作,並要求所屬24小時待命聽候指示,導致同仁們無時無刻無法得到喘息,精神上幾近崩潰,自己則早早下班,完全不用負責。至於本則貼文所涉及之司長身份,陳昭姿強調「已經交由邱泰源調查。」該貼文下有網友點名是霸凌者指的是心健司長,但當時的心健司長現已調往健保署任職。健保署長石崇良受訪表示,健保署目前也在了解署內霸凌情況,至於網友所提的情況,還會進一步瞭解。若如外界猜測,霸凌案件發生時不在健保署,且委員質詢內容已經交給部長,健保署會在部長有所指示時全力配合。此外,陳昭姿21日下午在臉書繼續針對衛福部霸凌醫師發文,透露:「當我質詢結束後,有衛福部的基層同仁跑來跟我助理說『謝謝委員主持公道』。剛才我本人也收到曾跟劉司長共事過的同仁傳訊表示,過去劉玉娟在擔任健保署台北業務組組長時,底下10幾個科長被她換光光,儼然是土皇帝!」她還表示,卓榮泰院長已經下令,所有部會一週內徹查內部霸凌案,「請邱泰源部長好好去了解衛福部的狀況,冰凍三尺非一日之寒。上午質詢時我也同時提出匿名粉專投訴衛福部另一個單位的案件,請邱部長一併徹查,有霸凌問題的恐怕不只疾管署和社保司。」

施政藍圖涵蓋多面向 張善政:提升建設打造桃園宜居永續城
桃園市長張善政30日於市議會第3屆第4次會期中,進行施政報告。報告內容涵蓋了「交通與公共安全」「政策與法規創新」、「青年教育與就業機會」、「婦幼健康與照護」等多個面向。未來,市府期待持續推動智慧城市建設,完善公共設施與服務等施政,致力打造宜居、永續發展的現代化城市。張善政表示,桃園在交通安全方面取得顯著進展,今年上半年交通事故與死傷案件數較去年同期下降近一成,這得益於通學廊道及道路設施的改善。此外,市府與體育署及球團合作,成功解決桃園棒球場逢雨必淹的問題,並於賽季空檔完成修繕,球場安全性獲得球員及教練的認可,讓球迷享受更舒適的觀賽體驗。針對近年來多起工廠及倉儲火災的發生,張善政指出,市府持續積極檢討並修訂現行法規,並呼籲中央同步修法。以修訂工廠輔導法為例,要求存放危險物品的場所需定期申報,同時修訂消防法,要求倉儲業者主動申報,以保障民眾安全。另外,有關青年關注如何在桃園就學、工作的相關議題,張善政說明,市府積極推動教育與就業政策,除了擴建及新建學校外,還舉辦國際夏令營,邀請國際專家學者為學生拓展視野。青年就業方面,市府則提供多樣實習機會,並輔導青農成立智慧農耕團,幫助青年就業。他也提及,市府在社會住宅方面也有顯著成就,已啟用4處社會住宅,另有13處正在興建或規劃中,將提供超過5,000戶,滿足市民的居住需求。值得注意的是,施政報告中也提及,市府推動多項婦女照護政策,如補助凍卵營養金、SMA篩檢及好孕專車等,打造完善的婦幼健康照護體系;同時,透過智慧科技提升城市安全與生活品質,榮獲多項國際獎項,成為智慧治理的典範。市府以「大眾運輸導向發展」(TOD)為核心,推動捷運綠線北段、橘線及青線的建設,逐步完善大眾運輸網絡,促進城市整體發展。最後,張善政強調,桃園正處於轉骨的關鍵階段,同時也是提升桃園各項建設與改善生活品質的好機會,過去一年多,市府團隊務實地推動許多政策,為桃園的未來打下穩固基礎。未來將掌握每一個提升桃園的機會,以更宏觀的視野打造符合未來發展趨勢的宜居城市。

降低新生兒死亡率 呼吸窘迫新藥9月納健保
台灣新生兒死亡率高,2022年為千分之2.8,高於OECD國家。為讓寶寶順利長大,健保署將「呼吸窘迫症候群」的新藥Curosurf納入給付,同時放寬Surfactant的用藥規定,呼吸困難的新生兒不必等到插管,就能使用藥物,預計9月上路,盼減少新生兒死亡憾事。早產兒常見的呼吸窘迫症候群,容易造成寶寶死亡,若能及時用藥,就能緩解呼吸急促、發紺症狀,減少插管的痛苦,甚至幫助寶寶脫離插管。自9月起,健保署將新藥Curosurf納入給付,用於呼吸窘迫的新生兒,受惠人數為245至659人,每人每年可省下4.4萬元。同時,Surfactant的給付規定也放寬,呼吸窘迫的新生兒不必等到插管,在使用連續性正壓呼吸器(CPAP),且氧氣需求大於3成的前提之下就能用藥,估計將有490至540人受惠,每次療程可省5.4萬元。為滿足SMA(脊髓性肌肉萎縮症)病友的期待,健保署也放寬含nusinersen成分的脊髓腔內注射藥物、含risdiplam成分的口服液劑的給付條件。從「3歲以下發病確診」擴增至「18歲以下發病確診」,同時取消上肢運動功能RULM大於等於15分的起始治療條件,預估每人每年藥費約660萬元,約250人受惠,新增19億元健保支出。目前臨床針對IL-36基因突變的全身性膿疱性乾癬尚無標靶治療藥物,健保署也透過暫時性支付方式納入支付。另也收載新藥risankizumab用於克隆氏症,並放寬含abrocitinib成分藥品用於12歲剄未滿18歲的中重度異位性皮膚炎者。這3種藥物的給付,分別在7到10月間上路。

8月新制懶人包!3次免費心理諮商「擴至45歲」 罕病SMA藥物「放寬給付」
目前時間已經來到7月底,在8月份,有多項重要政策、法規有進行變更,其中涵蓋了醫療、交通、教育、勞工環境等領域,像是衛福部擴大了免費心理諮商的對象範圍到45歲、罕見疾病脊髓性肌肉萎縮症(SMA)的藥物放寬給付,另外業主必須幫勞工提供適當的降溫設備,否則會面臨到罰款。綜合媒體報導指出,8月新制在醫療方面有兩項變動,一是衛福部擴大了免費心理諮商的對象範圍,從原本的15至30歲擴大到15至45歲,預計將有6萬人受惠,服務人次可達19萬。同時,健保署也宣布對脊髓性肌肉萎縮症(SMA)的藥物進行放寬給付,包含「背針、口服」2用藥,約280名病友將因此受益。交通方面,台鐵推出了新的電子票證常客回饋優惠,搭乘次數越多,優惠越大,最高可享受8折優惠。此外,台鐵還推出了30日和60日兩種電子定期票方案,為乘客提供更多選擇。教育領域部分,教育部公布了新的督導辦法,禁止在早自習、午休等時間進行考試,同時也規定國三升學模擬考不得在寒暑假結束後的第一周舉行。除此之外,高中職學生將獲得新的「身心調適假」,每學期最多可請3天。另外體育署也修改了體適能測試的內容,將「仰臥起坐」改為「仰臥捲腹」,並新增了「漸速耐力折返跑」項目。在勞工權益保護方面,勞動部修訂了職業安全衛生設施規則,要求雇主在高溫天氣下必須提供適當的降溫設備,否則將面臨3萬至30萬罰鍰。內政部則計劃在8月開始提供免費的住宅租賃法律諮詢服務,以協助租屋族維護自身權益。環境保護也是此次變更的重點之一。環境部宣布將於8月15日開始實施國內碳權交易及移轉管理辦法,為我國的碳排放交易市場奠定基礎。

全面放寬!SMA藥物納入健保給付 全台約400人受惠
罕病律師陳俊翰生前為爭取健保擴大給付脊髓性肌肉萎縮症(SMA)藥物奔走,健保署將全面放寬給付,包括脊椎注射劑和口服藥物,不再以「上肢運動功能分數」作為門檻,預計8月1日生效,全台約400人受惠,健保需支出20多億元。過去有SMA病友不得不赴陸求藥,如今終於可以回台治療,每年約可省下700多萬元藥費。SMA是一種罕見的神經退化性疾病,目前共有3種治療方式,包括脊椎注射劑、口服藥和基因治療,前兩者的健保給付條件相似,目前僅限上肢運動功能(RULM)大於15分者,100餘人獲得治療,但仍有250多人不適用健保給付。社團法人台灣生命之窗慈善協會理事長李怡潔表示,在全世界近40國已實現全給付的情況下,台灣病友仍然用生命等待救命藥,讓人倍感痛心與無奈。健保署醫審及藥材組長黃育文表示,本月20日共擬會同意取消運動功能(RULM)大於15分的評估條件,讓18歲以下的SMA患者都能接受脊椎注射劑或口服藥治療,預計8月1日生效,新增250名患者受惠,預計健保花費20多億元。黃育文說明,除了取消運動功能評估限制,如病人對於脊椎注射藥品有不耐受性,終身有一次機會可轉換為口服藥,但健保給付有下車評估機制,如果藥物對病人已經無效,就會停止給付。李怡潔轉述一位病友家屬感想,指健保嚴格的給付條件讓多數病友都被排除在外,面對孩子不斷退化的體況,不得不跨海350公里,到中國大陸尋求救命機會,經過幾次脊椎注射劑治療後,病況有明顯進步,不僅體力變好、站得更久,下樓梯也更穩,右手的運動功能更進步到滿分。一想到未來不需要再舟車勞頓跨海治療,很驚喜也充滿感恩。至於一劑要價4900萬元的SMA基因治療,目前健保採有條件給付,僅限出生6個月內發病者。黃育文表示,目前國內共有3人使用,本次共擬會並未討論放寬給付條件。

實習醫誤診釀16歲高中生慘死 日名古屋醫院院長鞠躬致歉
日本名古屋於2023年5月時發生一起醫療疏失事故,當地一名16歲男高中生因為腹痛前往日本紅十字會愛之醫療中心名古屋第二醫院就診,結果實習醫師誤判斷男高中生只是急性腸胃炎,便讓其取藥後返家。隔日,男高中生因為情況沒有好轉又再度就醫,結果被診斷出腸繫膜動脈症候群(SMA),雖然院方即時搶救,但男高中生還是在半個月後不幸逝世。而院方則在17日召開記者會公開致歉。根據日本媒體報導指出,紅十字會愛之醫療中心名古屋第二醫院表示,該名男高中生於2023年5月28日因為腹痛、嘔吐被緊急送到院內,後續實習醫師依據電腦斷層影像、抽血報告等結果,將其判斷為急性腸胃炎,讓男高中生領藥後就直接返家。但後續因為症狀沒有好轉,男高中生又再次被送回醫院檢查,這時才有其他醫師診斷出,男高中生所罹患的根本不是急性腸胃炎,而是腸繫膜動脈症候群。後續又因為延誤治療的關係,讓男高中生病情持續惡化,甚至在30日發生心肺停止的情況。醫院在搶救半個月後,於2023年6月15日宣告不治。院方表示,當時實習醫師在判斷男高中生的電腦斷層與脫水情況時有明顯失誤,結果導致將男高中生罹患的腸繫膜動脈症候群誤判為急性腸胃炎,最終讓男高中生因為延誤治療而不幸身亡。而在歷經一年的調查後,院方選擇在17日召開記者會,除了說明原由外,也對此次醫療事故公開致歉。除此之外,院方也強調,目前已設計了實習醫師可向資深醫師諮詢的制度,希望能以此避免再次發生類似情形。另外,目前院方也針對此事與死者家屬討論和解事宜。

New Balance推出夏日涼拖鞋系列,今年夏天穿拖鞋就是潮!
運動型的涼拖鞋流行起來!讓大家走在路上拖著走也很潮!New Balance也順應推出全新夏日涼拖鞋兩個系列,全系列各種新色應有盡有,從並搭配率性織帶設計打造夏日機能時尚,涼鞋更有輕量、抗污且防潑水,並使用柔軟增高氣墊中底支撐,除了讓腳底舒適,更休閒百搭!(圖/品牌提供) 戶外探索型格擔當,夏日機能涼拖鞋有點戶外探索風格的運動感設計,New Balance 750,是一款機能型涼拖鞋,本季推出兩款新色SUA750B3「復古紅」與SUA750C3「沉穩灰」,以機能織帶搭配細節包邊的寬版鞋面,增高柔軟鞋底的設計與波浪線條鞋床,加上鞋面飾有New Balance字樣點綴的魔鬼氈綁帶,提供更加舒適的腳感,後腳跟處還能輕鬆拆卸作為拖鞋使用。New Balance 750(中性款)/2,680元。(圖/品牌提供)踏青出遊風格首選,簡約流線涼鞋 帶有層次配色的600涼鞋系列,一次推出4款人氣新色,包含迷彩圖騰點對的SMA600B2「叢林棕」與SMA600C2「神秘藍」,以及品牌標誌性的SWA600B2「經典灰」與搭上近期熱門話題的Y2K風格SWA600F2「流星銀」。每個顏色都實穿又百搭,標誌性的 LOGO 彈性織帶、鞋跟的鬆緊帶設計與一體成形的鞋底提供穩定的包覆感與腳板的舒適支撐,絕對是都會時髦男女外出踏青、街頭遊走的風格首選。 New Balance 600(左上右上男款)(左下右下女款)/2,480元。(圖/品牌提供)

陳俊翰也因它所苦…全台300多人無藥可用 盼SMA藥物納健保
罕病律師陳俊翰日前疑因感冒併發肺部感染過世,對許多人來說,一場小感冒,只需吃藥幾天、多休息就會康復,但像陳俊翰這樣的脊髓性肌肉萎縮症(SMA)患者,進出醫院卻是日常。台灣生命之窗慈善協會理事長李怡潔曾因沒力氣把痰咳出,住院了1個多月才撿回一命,得靠救命藥挽回運動功能,但全台SMA患者中,仍有300多人被排除在給付門檻外,患者期盼政府能比照英、美等國,將SMA藥品全面開放,讓他們重拾功能改善的希望。北榮神經醫學中心周邊神經科主治醫師蕭丞宗說,SMA為隱性遺傳疾病,當父母一方缺乏SMN1基因,子女就有4分之1機率罹病,缺少該基因將導致運動功能減退,一般在幼兒期發現,但也有青少年、成年時期發現的案例,患者的運動功能會逐漸退化,甚至走入死亡。李怡潔去年因感冒出現肺炎,送至急診室時,第一時間就被問「要不要插管」,雖未入住加護病房,卻也住院了1個多月,使用了很多抗生素。她直言,自己感冒住院的經驗相當豐富,因沒有能力把痰咳出,一點感冒都需入院。遺憾的是,一些人進去後就再也沒機會出來。「病友不能再等。」李怡潔表示,台灣2020年將SMA藥物納入健保給付,2023年第2次叩關,3年的時間,30幾個國家已陸續開放,包括英、美、澳、羅馬尼亞,甚至在戰爭中的烏克蘭,都將SMA藥品全面納入給付,為何戰爭中的國家都那麼重視,台灣卻慢這麼多?目前SMA有3種藥物,受限於「上肢運動功能RULM≧15」與「3歲之前發病確診病歷證明」的嚴格給付門檻,國內400多名SMA患者中,仍有300多人看得到、用不到。蕭丞宗說,SMA病友即便是在年長者身上,用藥後都能達到改善的效果。以他的臨床經驗,就有49歲的患者用藥後步距增加、走路速度提高、動作加大,希望政府放寬給付條件,全面開放所有SMA患者使用。

台版「非常律師」陳俊翰過世 曾因SMA失去求生意志…棄高薪工作回台原因曝
位列民進黨不分區立委提名人的律師陳俊翰,兒時罹患脊髓性肌肉萎縮症(SMA)致使行動不便,而他先前因被脫口秀節目《賀瓏夜夜秀》的來賓、前中國大陸央視記者王志安於節目中談論,受到外界更多關注,怎料今(15)日傳出過世的消息,隨後民進黨證實,陳俊翰已於11日凌晨逝世,享年40歲。其實陳俊翰的病情一度讓他失去求生意志,好在母親謝季珍的陪伴在側,讓他決定堅持下去,後來更是放棄原本的美國高薪工作機會,回台推動罕見疾病及身心障礙相關政策,幫助弱勢群體發聲,勵志的生命經驗也讓他被外界封為台版「非常律師」。根據《ETtoday新聞雲》報導,陳俊翰早在1歲左右發現罹患脊髓性肌肉萎縮症,此病終將造成運動神經元喪失功能,進而肌肉無力、萎縮;在他就讀小六年級病情急轉直下,後來還被送進加護病房急救長達2個月,期間一度喪失求生意志,告訴母親「不要救我,我真的撐不下去了」,後來也因母親的長久陪伴,讓他及時轉念,有了堅持下去的動力。儘管陳俊翰失去雙腿,身體僅剩頭部、幾根手指頭還能自主活動,還遭告知若不用藥,存活時間僅剩3年至4年,但他高中畢業後仍考上台灣大學,雙主修會計系與法律系的課程,之後2度赴美取得法學博士學位,順利通過考試取得律師執照。不過陳俊翰選擇放棄原本的美國高薪工作機會、較為優越的治療環境回台,進入進入中研院法研所,也在政府部門推動罕見疾病及身心障礙相關政策,今年更是第11屆立法委員選舉,民進黨不分區名單中的身障提名人。他說,美國保險的機制強調「無差別、無歧視」,待在美國可以得到更為妥善的照顧與治療,反觀台灣卻有多達數十種罕病用藥尚未給付,因此盼能透過自身力量推動相關權益,「每一條生命都該被珍惜。」陳俊翰並稱,他覺得自己能有機會實現個人理想已經非常幸運,希望未來的台灣,所有的障礙者都不必受到先天條件的限制,「哪怕讓多一點人有機會接受用藥,也會比我自己在國外獲得治療更有價值。」後來長期鑽研人權、身心障礙政策與法律的他,更被譽為台版「非常律師」、「生命鬥士」。如今陳俊翰過世的消息一出,民進黨也以聲明回應,「陳俊翰律師因疑似感冒引起併發症,2/10前往台大醫院新竹分院急診,雖經醫護人員極力搶救,仍於2/11凌晨不幸逝世。我們聞訊感到無比震驚與悲傷,俊翰的家人低調辦理後事,年節期間未對外公布,今日委由民進黨黨中央代為發布消息。感謝各界關心,也請給家屬空間處理相關事宜。」

永遠無緣的立委 賴清德告白悼陳俊翰
民進黨本屆不分區立委候選人、知名人權律師陳俊翰,驚傳因感冒引起併發症送醫急救無效,大年初二2月11日凌晨不幸逝世於台大新竹分院,享年40歲,各界震驚。對此,準總統賴清德15日晚間於臉書表示,自己聽聞俊翰突然離世的消息感到非常哀痛,撐到年後,15日才敢聯繫陳俊翰母親,也希望能由民進黨協助治喪,送陳俊翰走完最後一程。賴清德感性告白,認為陳俊翰的身體限制不了夢想,大家將會永遠懷念俊翰陽光自信的生命精神。俊翰是天使走過人間,願祂安息,在天上看顧摯愛的土地,大家會帶著祂所堅持的理想價值,繼續前進。而前新竹市立委候選人、交大教授林志潔也在臉書表示,陳俊翰家居新竹,自己競選期間與其有許多接觸,15日下午也與陳媽媽通話,彼此在通話中都泣不成聲。無論如何,俊翰的勇敢,帶給很多人力量,讓大家知道,人生即使遇到困難,也能活得如此陽光。曾與陳俊翰因黨務密切互動的新北市議員林秉宥則感嘆「活著的人就是要背負著死去夥伴的願望繼續前進」,字裡行間盡是對好伙伴的不捨。陳俊翰是出生即診斷罹患「脊髓性肌肉萎縮症(SMA)」,全身僅剩頭部及幾根手指頭能出力,卻依然能在台灣大學會計與法律雙主修畢業後赴美求學,並取得法學博士學位、考取紐約州律師執照,在美原有大好前途,卻放棄高薪,回台推動罕病與身心障礙倡議。由於克服病痛創造奇蹟,又能如此大愛,被人稱為台版的「非常律師」。

陳俊翰猝逝!台大證實「肺部遭感染」 賀瓏發45字文哀悼:令人敬佩
民進黨不分區被提名人陳俊翰今(15日)離世,享年40歲。台大醫院新竹分院證實,陳俊翰因肺部感染病逝。日前才因來賓王志安發表歧視身障言論,而遭到輿論的《賀瓏夜夜秀》主持人賀瓏,也在晚間透過社群哀悼陳俊翰。民進黨表示,陳俊翰疑似因感冒引起併發症,於10日至台大醫院新竹分院急診,但仍不幸於11日凌晨逝世,家人低調處理後事,年節期間未對外公布,並委由黨中央代為發布消息,民進黨除感到無比震驚、悲傷,也請外界給家屬空間。台大新竹分院院長余忠仁則證實,陳俊翰入院急救後因肺部感染離世,但詳細內容需徵詢家屬是否同意說明。另《賀瓏夜夜秀》主持人賀瓏,在其所屬的的薩泰爾娛樂作出出「還在瞭解狀況中」簡短回應,於社群表示哀悼。賀瓏表示「對於陳俊翰律師的離世深感遺憾,陳律師面對生命堅韌的態度令人敬佩。感謝他對於臺灣的貢獻與付出」。此外,引爆歧視風暴的來賓、前央視記者王志安,也在推特刊出來不及傳達給陳俊翰的道歉信,表示「我的歉意再也無法抵達,真是令人無限的懊悔和遺憾」。《賀瓏夜夜秀》日前因王志安的言論被炎上。(圖/翻攝自YouTube/STR Network)陳俊翰為台灣首位罹患脊髓性肌肉萎縮症(SMA)的律師,前年取得美國密西根大學法學博士學位。生前任中研院法學研究所博士後研究學者,獲民進黨提名為排名第16位的不分區立委。與大眾熟知的漸凍症不同,SMA主要是在嬰兒及兒童發病的隱性遺傳疾病,患者會因運動神經元喪失功能而肌肉無力、萎縮,進而造成運動功能下降。陳俊翰生前除有「人權律師」美譽,更放棄國外治療機會,回台為罕病病友爭取平等醫療。

民進黨不分區立委提名人 陳俊翰律師驚傳過世
第11屆立法委員選舉,民進黨不分區名單中的身障提名人,位列第16名的陳俊翰律師,先前曾受到脫口秀節目《賀瓏夜夜秀》來賓,前大陸央視記者王志安討論,因此受到不少關注,如今卻傳出陳俊翰已過世的消息,讓人相當意外,但民進黨尚未發布該消息。據《鏡週刊》報導,民進黨不分區第16位名單的身障律師陳俊翰,傳出過世消息。陳俊翰先天患有「脊髓性肌肉萎縮症」罕見疾病SMA,全身只有眼睛、嘴巴以及一根小指頭可以活動。但他在家人的支持下,突破身體限制,考取律師高考榜首與會計師執照,並取得哈佛法學院碩士、密西根大學法學院博士,隨後進入中研院法學研究所,專精國際人權法、身心障礙政策與法律等研究。先前脫口秀節目《賀瓏夜夜秀》來賓,前大陸央視記者王志安討論他後引發爭議,他也發出聲明表示「很感謝賀瓏和夜夜秀團隊對於我和我所提出的理念的認同和支持,不過,他們大可不必勉強自己在2028一定要投給民進黨。」陳俊翰說,台灣的民主自由可貴之處,就是每個人都可以自由選擇自己認同、支持的政黨,沒有誰可以強迫誰,所以他們不必違背自己的心意,當然如果賀瓏及製作團隊在經過四年的人生歷練後,發現賴清德總統所領導的民進黨政府做得還不錯,而願意發自內心的支持,也非常歡迎投給民進黨。

陳俊翰籲各界關注罕病SMA用藥困境 盼政府重視病友權益
中國《央視》前記者王志安日前上《賀瓏夜夜秀》嘲諷罹患罕病的民進黨不分區立委提名人陳俊翰,引起輿論撻伐,節目團隊與主持人賀瓏對此道歉。陳俊翰律師30日出席脊髓性肌肉萎縮症(SMA)記者會時表示,對於賀瓏致歉已有完整回應,希望各界關注SMA患者的用藥困境,目前國內仍有300多位SMA患者不符合健保給付標準,他也是屬於「望藥興嘆」的一人,盼政府重視病友權益。陳俊翰自幼罹患罕見疾病SMA,用比別人加倍的努力,靠全身唯一能動的嘴巴控制輪椅和寫字,取得美國法學博士與律師執照,卻捨棄國外治療的機會,毅然返台為罕病病友爭取平等醫療。他坦言身體功能退化已是患者日常,但生命意義遠超出身體功能限制,靠著母親、旁人協助,自己還能從事人權倡議工作,努力完成夢想。陳俊翰表示,台灣罕見疾病患者有1萬7千多人,生活中面臨很多不同的困難,但在健保給付限制下,許多患者仍無藥可用。SMA藥物開放健保給付後,目前國內仍有300多人不符合健保給付標準,真正能使用藥物的患者大概僅有3分之1。他自己也是其中之一,僅接受症狀治療,無法阻止全身肌肉退化,只能望藥興嘆。不是漸凍症! SMA嬰兒死亡率最高疾病脊髓性肌肉萎縮症是一種因運動神經元退化,導致肌肉無力及萎縮的隱性遺傳性疾病。「台灣SMA之父」鐘育志教授表示,SMA與「漸凍症(ALS)」容易混淆,兩者雖然都與運動神經元退化有關,但漸凍症好發年齡介於50至75歲,SMA病友在18歲之內發病者高達99%。根據統計,台灣目前約有420名SMA病友,大人小孩皆有。SMA症狀包括肌肉張力低下、四肢與軀幹肌肉無力與肌肉萎縮,且運動功能隨著年齡增長而持續退化。鐘育志表示,坐輪椅的病友幾乎都是極重度肢體障礙者,最嚴重者除了肌肉沒力量,而且全身肌肉鬆軟到連自己的頭部垂倒時都無力撐起,不但照顧者難以抱起,病友可能連自己移位、如廁、刷牙、洗臉、拿碗、舉筷、咀嚼、吞嚥都有困難,甚至睡覺時翻身或呼吸的力氣也沒有,晚上還需使用呼吸器。SMA依發病年齡與最佳運動功能將疾病分為4種類型,其中約6成SMA病友在出生6個月以內發病,是最嚴重的第一型,陳俊翰便屬此型,若未能及早治療,2歲前死亡率高達8成,是全世界嬰兒死亡率最高的遺傳疾病。病程踩煞車 SMA治療有望讓運動功能進步台大醫院神經部教授蔡力凱表示,SMA過去無藥可醫,主要依據病人的需求提供營養與呼吸照護,幫助預防肌肉無力產生的併發症。直到2016年開始,SMA陸續出現俗稱的背針、基因治療與口服藥等3種新興治療藥物,透過影響特定基因表現,不但有助減緩惡化,「幫病程踩煞車」,真實世界的臨床觀察更發現及早治療有機會讓運動功能進步。SMA病友盼健保鬆綁年齡與門檻蔡力凱表示,全台約有420名SMA病友,健保給付治療已約百人受惠,但與時間賽跑的病友仍有近300人,這群病人主要因RULM(上肢運動功能)低於15分、目前功能狀態較嚴重,抑或是3歲以後發病,病程惡化速度相對較慢,而被健保拒於門外。根據文獻,即使目前功能狀態較嚴重,治療也有機會翻轉命運,且全球與國內數據也皆看到類似成果,呼籲政府給予每位SMA病友治療的機會。台灣首位SMA律師 盼病友享有平等醫療權益中華民國肌萎縮症病友協會法律顧問陳俊翰律師,不僅是現任中央研究院法律研究所博士後研究學者,更是台灣第一位前往美國取得法學博士與律師執照的SMA病友。雖然在美國擁有較好的治療機會與高薪,但他卻選擇回國,與SMA病友一同等待。陳俊翰認為,每一個生命都有與生俱來的意義,在海外求學時深刻體會到國際上對待人權的最基本尊重,就是珍惜他的生命價值,全球許多國家也對SMA治療採取全給付政策。至於台灣SMA病友須符合特定條件才能獲給付,陳俊翰坦言,從滿心希望藥物給付,到「有藥可用,但不包括我」的沮喪,這個等待過程與結果對病人來說是殘忍與煎熬的。每一位罕病病友都在跟時間賽跑,他們迫切需要藥物來阻止疾病惡化,這些藥不僅代表病友們是否可以活動,更代表生命的延續,每一條生命都有存在的意義,也都值得被救。台灣自詡人權立國,期待政府多關愛SMA與其他罕見疾病病友,享有應有的、平等的醫療權益,有機會發揮自我價值。

一吃就吐、體重不斷下降! 檢查竟是罕見「十二指腸阻塞」
9歲的小明1年多前開始無法正常進食,常常吃一點就覺得飽,甚至吃東西就會馬上有噁吐感,最近3個月症狀加劇,吃東西吐不停且體重驟降,從21公斤掉到18.5公斤!國泰綜合醫院小兒科主任級醫師林隆煌安排患者住院檢查,研判為十二指腸腸阻塞,罹患「上腸繫膜動脈症候群」,並指出此病若不進行治療,嚴重可能會致殘。十二指腸被夾住 又痛又噁心林隆煌表示,上腸繫膜動脈症候群(superior mesenteric artery syndrome)是罕見的上消化道阻塞疾病。當上腸繫膜動脈(SMA)壓迫腹主動脈(AA),會導致十二指腸內容物不能順利排入空腸,無法獲得適當的營養,導致體重減輕和營養不良。病因與後腹腔脂肪或結締組織流失有關,例如癌症、嚴重外傷、營養吸收不良症,或厭食症等,人變得太瘦、脂肪消失,使得腸子被血管夾住;在受傷或脊柱手術後被放置在身體石膏中的患者也會出現SMA綜合症,又稱為石膏症候群;另外也有解剖位置異常,使得十二指腸懸吊太高或上腸繫膜動脈起源處過低,都增加十二指腸被夾住的機率。▲上腸繫膜動脈症候群是罕見的上消化道阻塞疾病。(國泰綜合醫院提供)壓迫引起的疼痛會使人虛弱,引起「食物恐懼症」並加重病情。當十二指腸受壓會讓人噁心、嘔吐,當體重持續下降時,導致SMA和AA之間的角度減小,從而加重壓迫和阻塞。主動脈與上腸繫膜動脈的正常夾角約為38-56度,小明的夾角卻只有26.4度,壓迫到十二指腸,造成十二指腸阻塞及急性胃擴張。常見症狀包括噁心、嘔吐、腹痛、消化不良和早飽,儘管吃下的食物或飲料很少,但由於胃沒有排空,患者仍會感到飽,胃裡充滿了數小時前攝入的液體或食物,當胃排空延遲時會發生便秘,未消化的食物可能會嘔吐,當阻塞變得嚴重時可能會變成膽汁。攝入食物或飲料後腹痛可能會很嚴重,因為SMA的搏動變得更強並撞擊十二指腸。臨床症狀因人而異,有時症狀輕微,隨著時間推移會慢慢加重,如果不進行治療,某些人的症狀可能會嚴重致殘。噁心嘔吐腹痛消化不良早飽便秘致殘早期保守療法 嚴重需動手術林隆煌解說,治療方案包括藥物和手術干預,初期可以通過胃減壓、電解質校正和營養支持等保守療法進行管理。患有飲食失調症的人需要進行精神病學評估,當不能經口餵養時,可以通過放置在梗阻遠端的鼻空腸管進行腸內餵養,首先嘗試使用補充性高熱量液體進行頻繁的少量餵養,然後進行姿勢療法。接下來,從鼻子插入一根管子到空腸(鼻空腸管),特殊配方用於通過泵輸送16至20小時的餵養,提供患者所需的足夠卡路里和液體,這圍繞著通過鼻胃管以幫助實現胃和十二指腸減壓,通過靜脈輸液復甦,以及監測和更換血清電解質,經由全胃腸外營養以提供足夠的營養實現體重增加。而促運動劑有益於提升體重,對於病程較短的患者,保守治療最能解決上腸繫膜動脈症候群的症狀和體重,對於具有急性表現的兒科患者能受益於單獨的保守治療,體重增加的目標是恢復腸系膜脂肪墊和扭轉角度損失並減少AA和SMA之間的距離。如果保守措施在6至8週內無效並且症狀惡化,特別是患者有劇烈疼痛且無法耐受胃腸道餵養,則需要進行手術。目前最常用的是腹腔鏡十二指腸空腸吻合術,讓十二指腸的有效減壓,具有恢復快、腸蠕動功能改善和患者健康、小腸粘連和術後切口疝發生率降低、失血量最少、術後疼痛減輕以及最終美容效果好的優點。醫師最後提醒,上腸繫膜動脈症候群是腸梗阻的一種罕見原因,延誤診斷會影響患者的身心健康。早期診斷時可以對患者進行保守治療,但如果診斷延遲或藥物治療失敗,則需要手術干預。

治療看見曙光!罕病藥物納健保 健保署:已給付1病童4900萬
今年8月脊髓性肌肉萎縮症(Spinal Muscular Atrophy,簡稱SMA)基因治療藥物被健保署納入健保給付後,高雄1名男童持續接受高雄醫學大學附設中和紀念醫院治療,目前健保給付金額已高達4900萬。健保署署長石崇良表示,健保首次將罕病基因治療藥品納入給付,適應症為「6個月以下發病,且帶有基因突變」的SMA病人,預估1年有8至9名病童受惠。高雄1名4個月又11天大的男嬰出生後因頭部無法直立、全身軟弱無力,哭聲微弱,送到高醫看診,被診斷出為罕見地脊髓性肌肉萎縮症(Spinal Muscular Atrophy,簡稱SMA),在經過2個月的治療後頭部已經可以控制自如、扶著髖部可以坐立、手部抓握力量變強、並且能主動連續翻身、趴著時可以自主抬頭。而脊髓性肌肉萎縮症為一種漸進性神經肌肉退化性疾病,位於人類第五號染色體的SMN1基因缺損,導致脊髓前角細胞(運動神經元)退化,病人的運動功能會隨著時間,持續不斷的退化,導致全身肌肉無力、甚至無法自主呼吸而死亡。脊髓性肌肉萎縮症也是一種體隱性遺傳罕見疾病,必須要父母親都是帶有1個缺陷基因的帶因者,就會有1/4的機率生下SMA的孩子,但父母並不會發病。要正確診斷SMA除了上述臨床特徵外,需由小兒神經科或神經內科醫師依據臨床症狀、神經學檢查、肌電圖、SMN1及SMN2基因檢查,來做確診。石崇良表示,SMA現在已經有3種治療藥物,包括需終生給藥的RNA藥物(Nusinersen髓鞘內給藥)及小分子藥物(Risdiplam口服)和只需一次性給藥的基因治療藥物(Zolgensma靜脈注射),健保署自2020年7月起給付SMA脊髓注射藥物,2023年4月給付口服液劑,到2023年8月再將基因治療藥物納入給付,健保署逐步爭取預算逐步擴增給付範圍,目前對於此項給付已給付到4900萬元。高醫院長王照元表示,SMA發生率約為1/10000,最嚴重的第一型SMA病童,約80%在2歲前就會因呼吸衰竭死亡,是目前世界上嬰兒死亡率最高的遺傳疾病。健保給付第一個SMA基因治療個案已於8月23日在高醫執行藥物治療,家屬非常感謝政府與健保署同意SMA基因治療藥物由健保給付,讓孩子與其家屬免除每天面對孩子運動功能退化的恐懼及擔憂,可以改善寶寶的運動功能暨生活品質,重新燃起生命希望。

SMA病患仍有75%無法健保治療 協會推紀錄片盼大眾關注!
脊髓性肌肉萎縮症(SMA)是一種罕見疾病,過去無藥可醫,但現在共有3種藥物獲得健保有條件給付,讓患者獲得一絲希望。不過,台灣生命之窗慈善協會表示,目前仍有75%的SMA患者因不符合健保條件而無法使用健保治療。為了讓更多民眾了解SMA、關懷病友,台灣生命之窗慈善協會推出《這一刻,得來不易The Moment》紀錄片,並於9月14日舉辦首映記者會,邀請健保署、罕見疾病基金會、臺灣病友聯盟、台灣癌症基金會、YouTuber 77老大、資深媒體人羅友志、政黨代表等一同鼓勵病友追逐平凡夢想,堅持不放棄。罕病SMA運動功能退化 嚴重會影響呼吸、死亡SMA不僅是罕見疾病,也是一種退化性疾病,本身也是SMA病友的台灣生命之窗慈善協會理事長李怡潔表示,患者運動功能會逐漸退化,無法自理生活,嚴重會無法呼吸,甚至死亡。過去SMA無藥可醫,但現在治療有了進步,健保也擴大原本SMA藥物給付對象,並且將2種新藥納入給付。李怡潔理事長提到,聽到病友陸續傳來被核准使用且漸漸找回運動功能的好消息,心裡相當感動,但現在依然有三百多病友未獲健保給付。李怡潔理事長的母親也表示,過去得知女兒沒有藥可用很心疼,現在知道健保擴大給付的消息一方面很興奮,但另一方面也很難過,因為有些病友符合健保給付條件有藥可用,但有些病友沒有,現在病友正面臨有藥可醫,但沒有健保給付治療的機會,希望未來可以再把條件放寬,讓更多病友能獲得治療。仍有3百多病友等待治療 紀錄片盼給予鼓勵、支持根據台灣生命之窗慈善協會資料顯示,健保於今(2023)年4月起放寬SMA藥物給付後,超過20名病友已獲得治療,不過仍有75%仍無法獲得健保給付治療。健保署署長石崇良表示,生命無價,每個病人都有活下去的機會,健保一定會盡力依初衷照顧每位民眾。而台灣生命之窗慈善協會發表的《這一刻,得來不易》紀錄片中,也描述3名病友取得治療後,改變生命與全家生活的成果。希望鼓勵仍在等待治療的300多位SMA病友,也希望民眾持續關注SMA議題,李怡潔理事長表示,透過大眾的關注、支持期盼能盡早落實一個都不放棄。除了首度播映《這一刻,得來不易》紀錄片外,台灣生命之窗慈善協會也預告推出《迷霧前行》紀錄片,串連過去4支SMA家庭影片的未播出片段,傳遞SMA家庭對抗疾病的艱辛、團結、堅持與毅力,進而幫彼此加油打氣。

SMA成人患者30年來首獲治療機會 重拾未來信心與希望
【健康醫療網/記者王冠廷報導】「本以為一輩子就這樣了,沒想到過30歲還有治療的機會」,中國醫藥大學附設醫院神經部神經免疫暨基因疾病科主任 郭育呈醫師分享近期開始進行背針治療的30歲女性SMA患者心聲,在公家機關上班的她,在家人陪同下前來進行治療,「從5月底開始,目前進入第三次治療,非常期待透過治療有機會維持肌肉力量甚至有些微進步,這些平凡的小事對我跟家人來說都是值得慶祝的大事。」脊髓性肌肉萎縮症(簡稱SMA)為罕見遺傳性神經肌肉疾病,全國約400多位患者,因為脊髓及腦幹的運動神經元退化,導致嚴重持續性的肌肉萎縮、無力,最終導致病患行走或進食困難,甚至無法呼吸導致死亡。脊髓性肌肉萎縮症用藥給付範圍擴增 病友有機會不再退化郭育呈醫師說,SMA的藥物研發是醫療科技上很大的進展,及早使用藥物治療可以有效阻止疾病惡化,目前台灣有三個藥物上市,分別採背針、口服藥、基因治療等形式,納入健保的有背針與口服藥,顧名思義,這兩種治療藥物的給藥方式完全不同。背針是治療SMA的第一種用藥,將藥物直接送到中樞神經系統,也就是這個疾病發生的源頭,做最直接的治療,在全球有豐富的治療經驗,療效與安全性較明確。另一方面,口服是藥水的形式,必須冷藏,天天固定時間吃藥,跟其他口服藥一樣,需經腸道吸收發生作用,療效也有可能被血腦障壁影響。目前健保署是根據臨床實證和給付效益來決定擴增給付範圍,口服藥物可用在「2個月以上未滿18歲病患」,背針則是沒有年齡上的限制。郭育呈醫師進一步說明,今年健保署擴增SMA用藥給付範圍,對病友和醫界來說都是令人振奮的消息!意味著病友有機會用藥,有機會讓運動功能不再退化,接受治療對病友與其家庭都是改變生命的機會,病友得以保持基礎生活自理的能力,及至發揮自身所長回饋社會。非常欣慰看到這樣的進展與改變,感謝健保署照顧SMA病友。最後呼籲病友平時要照顧好自己的生理及心理健康,並可以盡快回診接受評估,確認自身是否符合用藥資格,一同邁向台灣SMA治療新篇章。

母親節最棒的禮物! 聽損媽靠基因檢測揭遺傳真相
本周日就是母親節,在大家苦惱母親節該送什麼禮物的時刻,一位新手媽媽迎來她最幸福的母親節。一直以來,身為客服人員的A小姐都有聽力不佳的困擾,看了許多間耳鼻喉科仍找不到原因。直到懷孕時,博愛蕙馨醫院婦產科曾翌捷醫師提醒她,可以做包含聽損的帶因篩檢,報告出來她才赫然發現,自己竟然是遺傳性聽損的患者和基因帶因者!根據國健署統計,台灣每1000個新生兒就有1個人雙耳聽障,其中2/3就是基因遺傳造成的,新生兒聽力篩檢每年更是發現超過700個聽損寶寶。由父母遺傳給孩子的突變基因引起的聽力損傷,研究發現至少數千種可能導致聽損的突變基因,有遺傳性聽損的新生兒,通常在出生時就出現聽力障礙,不但會阻礙語言學習,更會大大影響日後的生活。幸運的是,她的先生經過檢測,報告顯示沒有遺傳性聽損的隱性帶因,寶寶的遺傳患病機率因此大幅下降,夫妻倆才放心繼續懷孕生產。在孩子健康出生後,李小姐激動又開心地感謝曾醫師,不僅幫助她找到多年來聽力不好的原因,更重要的是,沒有讓遺傳疾病的遺憾,在寶寶的生命裡重演。曾翌捷醫師感嘆地說:「身為一位執業多年的婦產科醫師,看過許多夫妻明明沒有家族史,孩子出生後卻罹患了遺傳疾病,讓新手父母每每都措手不及。」因為遺傳疾病是無法透過國內現行的常規產檢(包含唐氏症篩檢、NIPT、羊膜穿刺的羊水核型分析、羊水晶片)來偵測,所以沒有家族史,就無法在產前針對某疾病做特定檢測。根據研究顯示,平均每個人身上帶有至少2個遺傳性疾病基因,這些遺傳性疾病,全球發生率約為1%,每當新生兒患病時,父母必然驚慌心痛。創源生技執行長蔡政憲進一步表示,遺傳疾病中,還包括X染色體脆折症(FXS)、脊髓性肌肉萎縮症(SMA)、藥害性聽損、等多種疾病,創源累積的160萬基因大數據顯示,在遺傳疾病帶因篩檢-孕知因的統計中發現,各種疾病的綜合帶因率高達50%,如果夫妻同時為同一疾病帶因者,孩子就有可能有1/4的機率患病,創源根據國際醫學會建議,提供基因致病機制明確、帶因率高、兒童時期發病且有後續治療或照護配套的檢測項目,幫助計劃懷孕的夫妻,或是已經懷孕的婦女及早篩檢,只要簡單抽1次血就可檢測。美國婦產科醫學會也建議,為了確認寶寶的患病風險,讓媽媽能安心待產,迎接健康新生兒,不分種族背景應該都要進行帶因篩檢。曾翌捷醫師再度強調,現今基因檢測的進步,已經讓這些不易被發現的隱性疾病,透過帶因篩檢被發現,輔助婦產醫師在有限的問診時間內,快速掌握病患的家族史、疾病帶因狀況,規劃適當的臨床照護,更能降低遺傳疾病的風險與負擔。